
The Not-alike3 command pipeline finds genomic targets for the molecular diagnosis of diseases caused by pathogen microorganisms
DOI:
https://doi.org/10.18633/biotecnia.v27.2495Keywords:
Python programming, Molecular diagnostics, Pathogen detection, Molecular biology, SHGAbstract
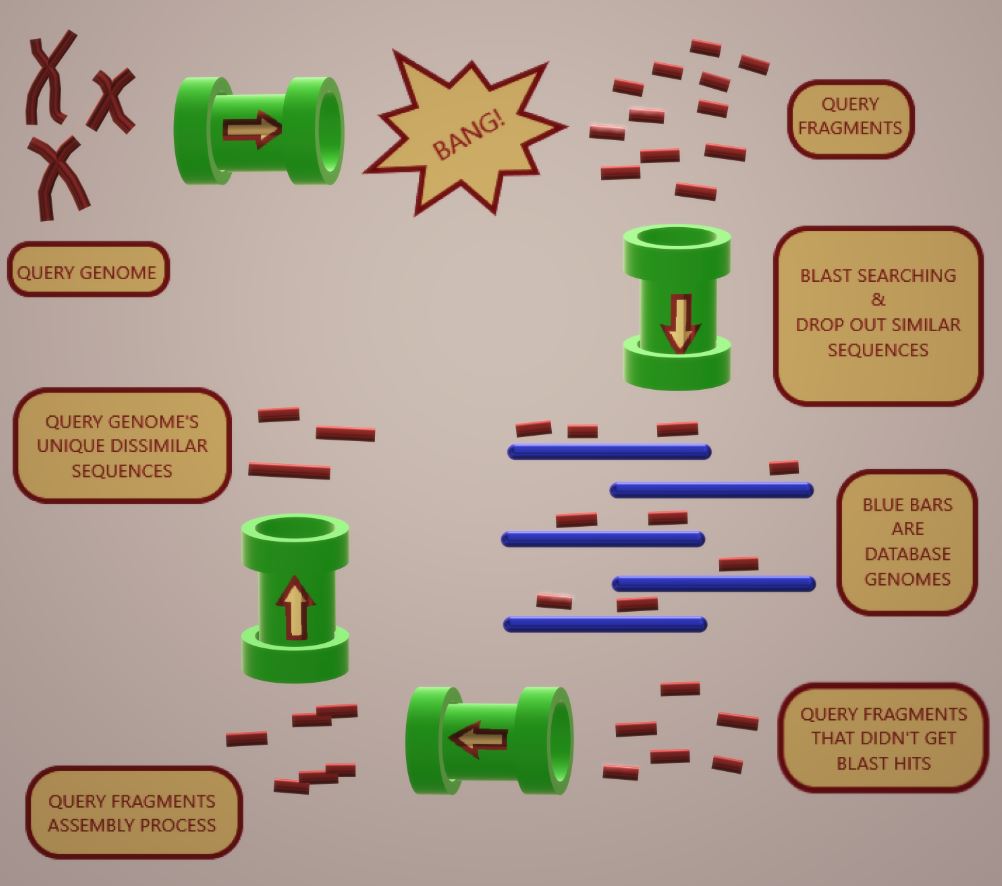
DNA-based molecular techniques are crucial for the precise identification of microorganisms. Despite their speed and sensitivity, specificity hinges on primer or probe design. The search for unique sequences in organisms’ genomes has various applications such as microorganism identification. There are published bioinformatic tools that employ the concept of in silico genomic subtractive hybridization analysis to identify species-specific/unique regions within a genome of interest. However, they use deprecated tools and program[1]ming languages that are not currently used in bioinformatics, some of them are specialized or difficult to obtain because the repositories, where these tools were hosted, are not public or the web servers are currently down. To address these issues, we implemented the in silico genomic subtractive hybridization idea in a user-friendly, open-source, freely available, easy-to-install command pipeline application written in Python (called Not-alike3). We designed PCR primers over the sequence of unique regions identified in the genomes of two Mucorales species, Rhizopus oryzae and Cuninghamlla bertholletiae, employing Not-alike3 command pipeline; we made specificity tests to challenge these primers and we observed that they were species-specific.
Downloads
References
Argimón, S., Konganti, K., Chen, H., Alekseyenko, A.V., Brown, S. and Caufield, P.W. 2014. Comparative genomics of oral isolates of Streptococcus mutans by in silico genome subtraction does not reveal accessory DNA associated with severe early childhood caries. Infection, Genetics and Evolution. 21: 269-278. DOI: https://doi.org/10.1016/j.meegid.2013.11.003.
Baldin, C., Soliman, S.S., Jeon, H.H., Alkhazraji, S., Gebremariam, T., Gu, Y., Bruno, V.M., Cor-nely, O.A., Leather, H.L., Sugrue, M.W., Wingard, J.R., Stevens, D.A., Edwards, J.E. and Ibra-him, A.S. 2018. PCR-based approach targeting mucorales-specific gene family for diagnosis of mucormycosis. Journal of clinical microbiology. 56(10): 1110-1128. DOI: https://doi.org/10.1128/jcm.00746-18.
Barh, D., Tiwari, S., Jain, N., Ali, A., Santos, A.R., Misra, A.N., Azevedo, V. and Kumar, A. 2011. In silico subtractive genomics for target identification in human bacterial pathogens. Drug Development Research. 72(2): 162-177. DOI: https://doi.org/10.1002/ddr.20413.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K. and Madden, T.L. 2009. BLAST+: architecture and applications. BMC bioinformatics. 10: 1-9. DOI: https://doi.org/10.1186/1471-2105-10-421.
Chetouani, F., Glaser, P. and Kunst, F. 2001. FindTarget: software for subtractive genome analy-sis. Microbiology. 147(10): 2643-2649. DOI: https://doi.org/10.1099/00221287-147-10-2643.
Davi, M.J.P., Jeronimo, S.M.B., Lima, J.P.M.S. and Lanza, D.C.F. 2021. 2021. Design and in silico validation of polymerase chain reaction primers to detect severe acute respiratory syndrome coro-navirus 2 (SARS-CoV-2). Scientific Reports. 11: 12565 https://doi.org/10.1038/s41598-021-91817-9.
Gardès, J., Croce, O. and Christen, R. 2012. In silico analyses of primers used to detect the patho-genicity genes of Vibrio cholerae. Microbes Environ. 27(3): 250-6. DOI: https://doi.org/10.1264/jsme2.me11317.
Haubold, B., Klötzl, F., Hellberg, L., Thompson, D. and Cavalar, M. 2021. Fur: Find unique ge-nomic regions for diagnostic PCR. Bioinformatics. 37(15): 2081-2087. doi: https://doi.org/10.1093/bioinformatics/btab059.
Kim, D., Paggi, J.M., Park, C., Bennett, C. and Salzberg, S.L. 2019. Graph-based genome align-ment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology. 37(8): 907-915. DOI: https://doi.org/10.1038/s41587-019-0201-4.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and 1000 Genome Project Data Processing Subgroup. 2009. The sequence align-ment/map format and SAMtools. Bioinformatics. 25(16): 2078-2079. DOI: https://doi.org/10.1093/bioinformatics/btp352.
Liu, Q., Jin, X., Cheng, J., Zhou, H., Zhang, Y. and Dai Y. 2023. Advances in the application of molecular diagnostic techniques for the detection of infectious disease pathogens (Review). Mol Med Rep. 27(5):104. DOI: https://doi.org/10.3892/mmr.2023.12991.
O’Leary, N.A., Cox, E., Holmes, J.B., Anderson, W.R., Falk, R., Hem, V., Tsuchiya, M.T.N., Schuler, G.D., Zhang, X., Torcivia, J., Ketter, A., Breen, L., Cothran, J., Bajwa, H., Tinne, J., Meric, P.A., Hlavina, W. and Schneider, V.A. 2024. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Sci Data. 11(1): 732. DOI: https://doi.org/10.1038/s41597-024-03571-y
Pagès, H., Aboyoun, P., Gentleman, R. and DebRoy, S. 2024. Biostrings: Efficient manipulation of biological strings. Bioconductor R version package 2.70.2. DOI: https://doi.org/doi:10.18129/B9.bioc.Biostrings.
Pertea, G. and Pertea, M. 2020. GFF utilities: GffRead and GffCompare. F1000Research. 9. DOI: https://doi.org/10.12688%2Ff1000research.23297.2.
Pertea, M., Kim, D., Pertea, G.M., Leek, J.T. and Salzberg, S.L. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature protocols. 11(9): 1650-1667. DOI: https://doi.org/10.1038/nprot.2016.095.
Portela, J., Grunau, C., Cosseau, C., Beltran, S., Dantec, C., Parrinello, H. and Boissier, J. 2010. Whole-genome in-silico subtractive hybridization (WISH)-using massive sequencing for the identi-fication of unique and repetitive sex-specific sequences: the example of Schistosoma mansoni. BMC genomics. 11: 1-8. DOI: https://doi.org/10.1186/1471-2164-11-387.
Shao, Y., He, X., Harrison, E.M., Tai, C., Ou, H.Y., Rajakumar, K. and Deng, Z. 2010. mGe-nomeSubtractor: a web-based tool for parallel in silico subtractive hybridization analysis of multiple bacterial genomes. Nucleic Acids Research. 38: W194-W200. DOI: https://doi.org/10.1093/nar/gkq326.
Singh, V. and Mishra, R.K. 2010. RISCI-Repeat Induced Sequence Changes Identifier: a com-prehensive, comparative genomics-based, in silico subtractive hybridization pipeline to identify re-peat induced sequence changes in closely related genomes. BMC bioinformatics. 11: 1-25. DOI: https://doi.org/10.1186/1471-2105-11-609.
Ueda, S., Washio, K. and Kurosaki, K. 1990. Human-specific sequences: Isolation of spe-cies-specific DNA regions by genome subtraction. Genomics. 8(1): 7-12. DOI: https://doi.org/10.1016/0888-7543(90)90219-K.
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B.C., Remm, M. and Rozen, S.G. 2012. Primer3 – New capabilities and interfaces, Nucleic Acids Research. 40(15): e115 – e115. DOI: https://doi.org/10.1093/nar/gks596.
van Weezep, E., Kooi, E.A. and van Rijn, P.A. 2019. PCR diagnostics: In silico validation by an automated tool using freely available software programs. J Virol. Methods. 270: 106-112. DOI: https://doi.org/10.1016/j.jviromet.2019.05.002.
Vidic, J., Manzano, M., Chang, C.M. and Jaffrezic-Renault, N. 2017. Advanced biosensors for detection of pathogens related to livestock and poultry. Veterinary Research. 48(1): 1-22. DOI: https://doi.org/10.1186/s13567-017-0418-5.
Voigt, K., Cigelnik, E. and O'donnell, K. 1999. Phylogeny and PCR identification of clinically important Zygomycetes based on nuclear ribosomal-DNA sequence data. Journal of Clinical Micro-biology. 37(12): 3957-3964. DOI: https://doi.org/10.1128/jcm.37.12.3957-3964.1999.
Wang, X., Fu, Y.F., Wang, R.Y., Li, L., Cao, Y.H., Chen, Y.Q., Zhao, H.Z., Zhang, Q.Q., Wu, J.Q., Weng, X.H., Cheng, X.J. and Zhu, L.P. 2014. Identification of clinically relevant fungi and prototheca species by rRNA gene sequencing and multilocus PCR coupled with electrospray ioni-zation mass spectrometry. PLoS One. 9(5): e98110. DOI: https://doi.org/10.1371/journal.pone.0098110.
Downloads
Published
How to Cite
Issue
Section
License
Copyright (c) 2025

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.

The journal Biotecnia is licensed under the Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0) license.





_(2).jpg)




